
Rare Types of Anemia
What is anemia?
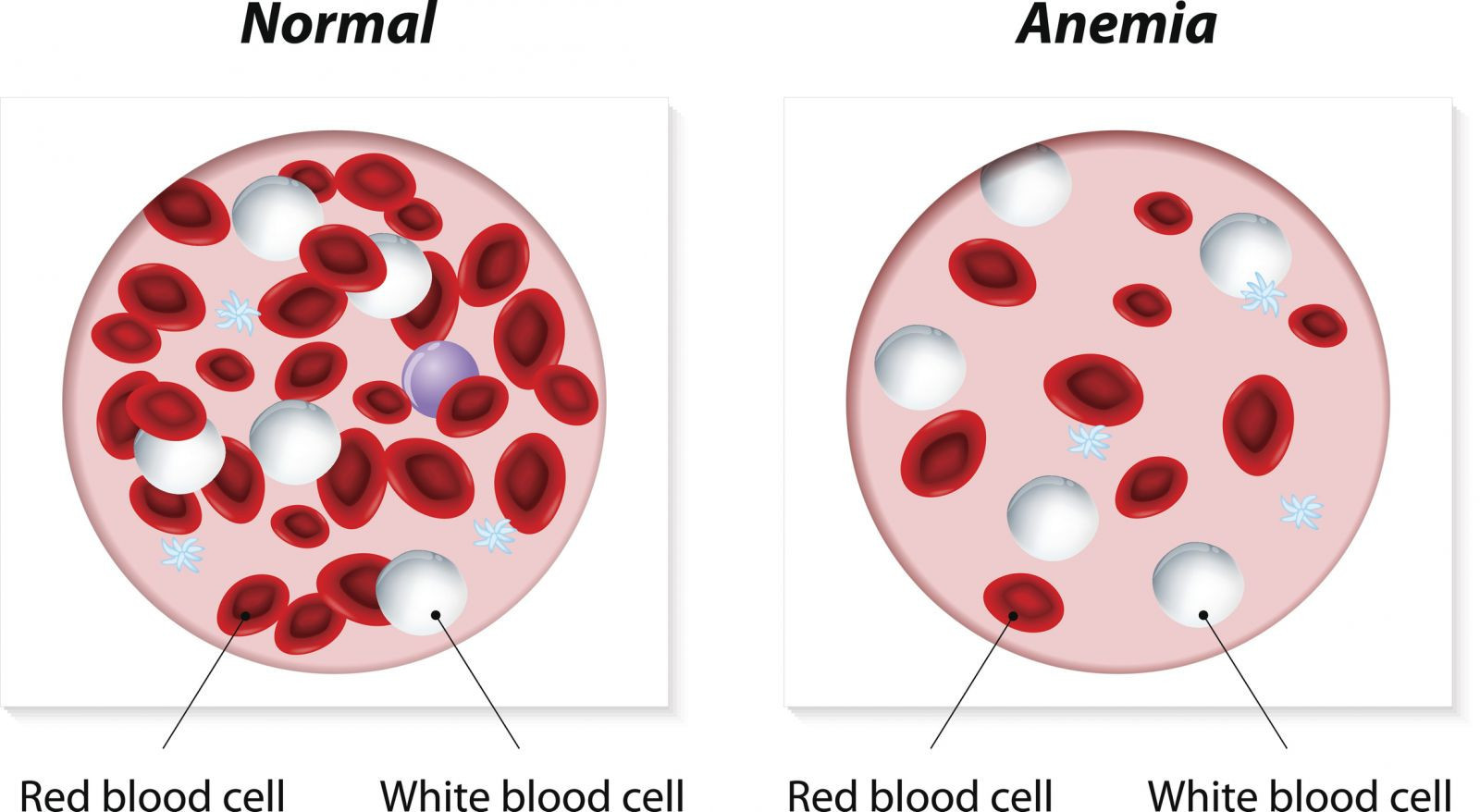
Anemia is a medical condition characterized by a deficiency of red blood cells or hemoglobin in the blood. Red blood cells and hemoglobin are responsible for carrying oxygen from the lungs to the rest of the body. When levels of red blood cells or hemoglobin are low, the body may not get enough oxygen, leading to symptoms such as fatigue, weakness, and shortness of breath.
There are several types of anemia, each with its own causes and treatments. Some common types of anemia include:
- Iron deficiency anemia: This is the most common type of anemia and is caused by a lack of iron in the body, which is needed to produce hemoglobin.
- Vitamin deficiency anemias: Anemia can also be caused by a deficiency in vitamins such as vitamin B12 or folate, which are necessary for red blood cell production.
- Anemia of chronic disease: This type of anemia is associated with chronic conditions such as chronic kidney disease, cancer, or inflammatory disorders, which can interfere with the body’s ability to produce red blood cells.
- Hemolytic anemias: These are a group of anemias caused by the premature destruction of red blood cells, which can be due to inherited conditions, autoimmune disorders, or infections.
- Aplastic anemia: This rare but serious condition occurs when the bone marrow fails to produce enough red blood cells, white blood cells, and platelets.
Symptoms of anemia can vary depending on the underlying cause and the severity of the condition but may include fatigue, weakness, pale skin, shortness of breath, dizziness, headaches, and cold hands and feet. Treatment for anemia depends on the cause but may include dietary changes, iron or vitamin supplements, medications, or, in severe cases, blood transfusions or other procedures.
What is aplastic (or hypoplastic) anemia?
Aplastic anemia is a rare but serious condition in which the bone marrow fails to produce enough red blood cells, white blood cells, and platelets. Bone marrow is the spongy tissue inside bones where blood cells are produced. In aplastic anemia, the bone marrow is replaced with fatty tissue, leading to a reduction in the production of blood cells.
The exact cause of aplastic anemia is often unknown, but it can be acquired or inherited. Acquired aplastic anemia is more common and can be triggered by exposure to certain drugs, chemicals, or toxins, as well as viral infections, autoimmune disorders, and certain cancers. Inherited forms of aplastic anemia are usually due to genetic mutations that affect the bone marrow.
Symptoms of aplastic anemia can vary but may include fatigue, weakness, pale skin, shortness of breath, dizziness, headaches, and frequent infections. In severe cases, aplastic anemia can lead to life-threatening complications such as bleeding and infections.
Treatment for aplastic anemia depends on the underlying cause and the severity of the condition but may include medications to stimulate the production of blood cells, blood transfusions to replace deficient blood cells, and bone marrow transplant in severe cases. With proper treatment, many people with aplastic anemia can lead normal lives, although long-term monitoring and care are often needed.
What is sideroblastic anemia?
Sideroblastic anemia is a type of anemia characterized by the presence of abnormal iron deposits in the red blood cells. These iron deposits, called siderosomes, can be seen under a microscope and are a hallmark of the condition. Sideroblastic anemia can be inherited or acquired and can be classified as either congenital or acquired.
- Congenital sideroblastic anemia: This type of sideroblastic anemia is rare and is usually inherited. It is caused by genetic mutations that affect the production of heme, a component of hemoglobin. Without heme, the iron that is normally used to make hemoglobin accumulates in the red blood cells, leading to the formation of siderosomes.
- Acquired sideroblastic anemia: This type of sideroblastic anemia is more common and can be caused by a variety of factors, including:
- Vitamin B6 deficiency: Vitamin B6 is necessary for the proper function of enzymes involved in heme synthesis. A deficiency of vitamin B6 can lead to the accumulation of iron in the red blood cells.
- Alcohol abuse: Chronic alcohol consumption can interfere with the absorption and metabolism of vitamin B6, leading to sideroblastic anemia.
- Lead poisoning: Lead can inhibit enzymes involved in heme synthesis, leading to the accumulation of iron in the red blood cells.
Symptoms of sideroblastic anemia can vary but may include fatigue, weakness, pale skin, shortness of breath, dizziness, and an enlarged spleen. Treatment for sideroblastic anemia depends on the underlying cause but may include vitamin B6 supplements, medications to reduce iron overload, and blood transfusions in severe cases.
What are myelodysplastic syndromes?
Myelodysplastic syndromes (MDS) are a group of disorders characterized by abnormal development and maturation of blood cells in the bone marrow. In MDS, the bone marrow fails to produce enough healthy blood cells, leading to low levels of red blood cells, white blood cells, and platelets. The exact cause of MDS is often unknown, but it is thought to be related to genetic mutations in the blood-forming cells.
MDS can be classified into several subtypes based on the specific blood cell affected and the degree of dysplasia (abnormal cell development) present. The main types of MDS include:
- Refractory anemia: This is the most common subtype of MDS and is characterized by a deficiency of red blood cells, leading to anemia.
- Refractory cytopenia with multilineage dysplasia: This subtype is characterized by low levels of multiple types of blood cells (pancytopenia) and abnormal cell development in multiple cell lines.
- Refractory anemia with excess blasts: This subtype is characterized by an excess of immature blood cells (blasts) in the bone marrow and blood.
- Refractory cytopenia with unilineage dysplasia: This subtype is characterized by low levels of a single type of blood cell (such as red blood cells or platelets) and abnormal cell development in that cell line.
Symptoms of MDS can vary but may include fatigue, weakness, shortness of breath, pale skin, easy bruising or bleeding, and frequent infections. Treatment for MDS depends on the subtype and severity of the disease but may include supportive care such as blood transfusions or growth factors to stimulate blood cell production, medications to suppress the immune system, and in some cases, stem cell transplant.
What are autoimmune hemolytic anemia?
Autoimmune hemolytic anemia (AIHA) is a rare condition in which the body’s immune system attacks its own red blood cells, leading to their destruction. This results in a shortage of red blood cells, which are needed to carry oxygen to the body’s tissues. AIHA can be classified as either warm antibody hemolytic anemia or cold agglutinin disease, depending on the type of antibodies involved and the temperature at which they are active.
- Warm antibody hemolytic anemia: This is the most common form of AIHA and occurs when the immune system produces antibodies that attack red blood cells at normal body temperature (37°C or 98.6°F). This type of AIHA is often associated with underlying autoimmune disorders, such as lupus or rheumatoid arthritis, or can occur without an underlying cause (idiopathic).
- Cold agglutinin disease: This form of AIHA occurs when the immune system produces antibodies that attack red blood cells at lower temperatures, typically below normal body temperature. This can happen in response to infections, such as Mycoplasma pneumoniae or infectious mononucleosis, or in association with other conditions, such as lymphoma or chronic lymphocytic leukemia.
Symptoms of AIHA can vary but may include fatigue, weakness, pale skin, jaundice (yellowing of the skin and eyes), dark urine, and an enlarged spleen. Treatment for AIHA depends on the underlying cause and severity of the condition but may include corticosteroids to suppress the immune system, immunosuppressive medications, blood transfusions, and in some cases, removal of the spleen (splenectomy).
What is congenital dyserythropoietic anemia?
Congenital dyserythropoietic anemia (CDA) is a group of rare genetic disorders characterized by abnormal development of red blood cells in the bone marrow. This results in ineffective erythropoiesis, meaning that the bone marrow produces red blood cells that are not fully functional. There are several types of CDA, each caused by mutations in different genes involved in red blood cell production.
The main types of CDA include:
- CDA type I: This is the most common form of CDA and is characterized by abnormalities in the structure of red blood cells, leading to their premature destruction (hemolysis).
- CDA type II: This form of CDA is characterized by abnormal iron deposits in the red blood cells, which can lead to iron overload in the body.
- CDA type III: This is a rare form of CDA that is characterized by mild to moderate anemia and is often associated with other health problems, such as skeletal abnormalities.
Symptoms of CDA can vary but may include anemia, fatigue, weakness, pale skin, jaundice (yellowing of the skin and eyes), and an enlarged spleen. Treatment for CDA depends on the type and severity of the condition but may include blood transfusions, iron chelation therapy to reduce iron overload, and medications to support red blood cell production. In severe cases, a bone marrow transplant may be necessary to replace the defective bone marrow with healthy donor cells.
What is Diamond-Blackfan anemia?
Diamond-Blackfan anemia (DBA) is a rare genetic disorder characterized by a failure of the bone marrow to produce an adequate number of red blood cells. This results in a low red blood cell count (anemia), which can lead to symptoms such as fatigue, pale skin, and an increased risk of infections.
DBA is typically diagnosed in infancy or early childhood, although it can sometimes be diagnosed later in life. The exact cause of DBA is often unknown, but it is thought to be related to mutations in genes that are involved in the production of red blood cells.
In addition to anemia, individuals with DBA may also have other physical abnormalities, such as abnormalities of the face, head, hands, or heart. These abnormalities can vary widely among affected individuals.
Treatment for DBA usually involves regular blood transfusions to maintain a normal red blood cell count. In some cases, medications such as corticosteroids may be used to stimulate red blood cell production. In severe cases, a bone marrow transplant may be necessary to replace the defective bone marrow with healthy donor cells.
While there is currently no cure for DBA, with appropriate treatment, many individuals with the condition are able to lead relatively normal lives. Regular monitoring and management by a healthcare team familiar with DBA are important to ensure the best possible outcome.
What is megaloblastic anemia?
Megaloblastic anemia is a type of anemia characterized by the presence of unusually large, structurally abnormal, and immature red blood cells called megaloblasts in the bone marrow. These abnormal red blood cells do not function properly, leading to a decrease in the number of mature red blood cells and a decrease in the oxygen-carrying capacity of the blood.
The most common cause of megaloblastic anemia is a deficiency in vitamin B12 or folate (vitamin B9). Both of these vitamins are necessary for the production of DNA and for normal red blood cell maturation. Without adequate levels of vitamin B12 or folate, red blood cells cannot divide properly, leading to the formation of megaloblasts.
Symptoms of megaloblastic anemia can vary but may include fatigue, weakness, pale skin, shortness of breath, dizziness, and an enlarged liver or spleen. In severe cases, megaloblastic anemia can lead to neurological symptoms, such as numbness and tingling in the hands and feet, difficulty walking, and memory problems.
Treatment for megaloblastic anemia involves correcting the underlying vitamin deficiency. This may include vitamin B12 injections or oral supplements, or folic acid supplements, depending on the specific deficiency. In some cases, underlying conditions that may be causing the deficiency, such as malabsorption disorders, may also need to be treated.
What is Fanconi anemia?
Fanconi anemia is a rare genetic disorder that primarily affects the bone marrow, resulting in decreased production of all types of blood cells. It is named after the Swiss pediatrician who first described it, Guido Fanconi.
The condition is characterized by a range of physical abnormalities, bone marrow failure, and an increased risk of developing certain cancers, particularly leukemia and solid tumors. Fanconi anemia is inherited in an autosomal recessive manner, meaning that a person must inherit two mutated copies of the responsible gene (one from each parent) to develop the disorder.
Symptoms of Fanconi anemia can vary widely but may include:
- Anemia: Low red blood cell count, leading to fatigue, weakness, and pale skin.
- Thrombocytopenia: Low platelet count, leading to an increased risk of bleeding.
- Leukopenia: Low white blood cell count, leading to an increased risk of infections.
- Physical abnormalities: These may include skeletal abnormalities, skin discoloration, and abnormalities of the kidneys, heart, or other organs.
- Increased cancer risk: Individuals with Fanconi anemia have a significantly increased risk of developing leukemia, solid tumors (such as head and neck cancers), and certain other cancers.
Treatment for Fanconi anemia is aimed at managing symptoms and complications. This may include blood transfusions to treat anemia and medications to stimulate the production of blood cells. Bone marrow transplant may be considered in some cases to replace the defective bone marrow with healthy donor cells. Early diagnosis and regular monitoring by a healthcare team familiar with Fanconi anemia are important to manage the condition and reduce the risk of complications.




{kind=link}
{kind=link}
{kind=link}
{kind=link}